
Policitemia vera
La policitemia vera (conosciuta anche come "policitemia rubra vera" o "policitemia primaria" o semplicemente "policitemia") è un disordine mieloproliferativo cronico caratterizzato da una produzione abnorme di globuli rossi.
Il termine "policitemia" indica un aumento del numero di cellule, in particolare di globuli rossi (come suffragato dall'aggettivo latino "rubra", che significa, appunto, rossa).
I suffissi "vera" o "primaria" servono a differenziare questa patologia dalle policitemie secondarie, ovvero delle condizioni, non necessariamente patologiche, in cui l'organismo cerca di compensare un deficit cronico di ossigenazione tissutale con un aumento del numero dei globuli rossi, come avviene nei soggetti che vivono in alta quota, nei forti fumatori, in alcune malattie cardiache e polmonari.
Invece, la policitemia vera è una proliferazione neoplastica (cioè un tumore) di tipo clonale (cioè tutte le cellule tumorali derivano da un'unica cellula progenitrice localizzata nel midollo osseo, che perde il controllo ed inizia a proliferare), senza che vi sia alcuno stimolo esterno.
Insieme a leucemia mieloide cronica, mielofibrosi idiopatica e trombocitemia essenziale, la policitemia rubra vera appartiene al gruppo delle cosiddette sindromi mieloproliferative croniche.
Nel resto dell'articolo si discuterà della policitemia vera e ci si riferirà ad essa con l'acronimo PV.
Epidemiologia
La PV è una patologia piuttosto rara, con un'incidenza di circa 2 nuovi casi ogni 100.000 persone all'anno.
Similmente alla maggior parte delle patologie tumorali, tende a colpire maggiormente gli individui più in là con gli anni, con un picco di incidenza intorno ai 70 anni e una leggere prevalenza nel sesso maschile.
Eziopatogenesi
Non è nota una causa univoca che sia in grado di provocare la PV, ma, come per molte altre neoplasie, un aumento dell'esposizione ad agenti mutageni (in particolare, le radiazioni ionizzanti) rappresenta un importante fattore di rischio.
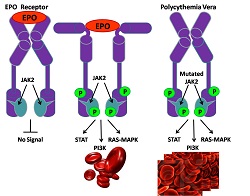
Dal punto di vista molecolare, la PV è contraddistinta in più del 95% dei casi da una mutazione acquisita di una proteina chinasi denominata "JAK2".
JAK2 è una proteina coinvolta nella via di trasduzione del segnale mediata dall'eritropoietina (più nota con l'acronimo "EPO", una sostanza tristemente assurta agli onori della cronaca per questioni legate al doping), un ormone prodotto dal rene che stimola la produzione di globuli rossi.
Nei pazienti affetti da PV, la mutazione di JAK2 rende la chinasi costituzionalmente attiva, indipendentemente dal segnale innescato dall'eritropoietina e, per questo, si verifica una proliferazione indiscriminata delle cellule midollari della serie rossa. In realtà, possono risultare aumentate anche le piastrine e alcuni tipi globuli bianchi, ma, comunque, lo stipite cellulare nettamente prevalente sono i globuli rossi.
Sintomatologia
In una discreta percentuale di casi, la PV può essere asintomatica e passare inosservata per molto tempo.
Nelle forme manifeste, è di frequente riscontro un intenso prurito, caratteristico perché tende ad accentuarsi in seguito ad un bagno caldo. La ragione di questo fenomeno non è nota con certezza, ma sembra risiedere in una iperproduzione di istamina e/o di prostaglandine.
Inoltre, l'eccesso di istamina può determinare anche lo sviluppo o il peggioramento di un'ulcera peptica.
A causa dell'aumentato ricambio cellulare, si verifica un aumento del tasso ematico di acido urico (l'acido urico è un prodotto di scarto del catabolismo del DNA), con incremento del rischio di attacchi di gotta e di nefrolitiasi.
Molto spesso è presente un'importante splenomegalia, ovvero un incremento di volume della milza, che a sua volta può essere causa di senso di tensione in addome, nausea e sazietà precoce.
Il rischio più grave connesso alla PV è l'aumento dell'incidenza di trombosi sia venosa che arteriosa, che può comportare eventi infausti come infarti, ictus, trombosi venose profonde e trombosi delle vene sovraepatiche. Ciò si spiega col fatto che, banalmente, più elementi cellulari ci sono in un vaso, maggiore è la viscosità e quindi la probabilità che esso si ostruisca.
Un altro fenomeno peculiare (ma presente anche in altre sindromi proliferative) è la cosiddetta "eritromelalgia", ovvero la comparsa di improvvisi dolori urenti (cioè che bruciano) alle mani e/o ai piedi, accompagnati da una colorazione rosso-bluastra della cute, in corrispondenza delle zone dolenti.
Infine, alcuni soggetti sviluppano un aspetto estetico particolare, denominato "pletora" e caratterizzato da colorito rosso intenso (o talvolta bluastro) della cute e delle membrane congiuntivali.
Dal punto di vista delle condizioni generali, come in molte altre neoplasie, sono frequenti dolori ossei, scarso appetito, calo ponderale, deficit di forza, mancanza di concentrazione e mal di testa.
Diagnosi
La sintomatologia appena descritta, se presente, induce ad effettuare una serie di accertamenti diagnostici.
Lo stesso avviene per le forme asintomatiche, allorché un semplice esame emocromocitometrico dimostri dei segnali d'allarme.
L'alterazione più caratteristica ed evidente della PV è l'aumento esponenziale del numero di globuli rossi, che possono raggiungere i 10.000.000 per microilitro (i valori normali sono di circa 5.000.000 per microlitro). Parallelamente, si verifica un importante incremento dell'emoglobina e dell'ematocrito (ovvero il rapporto tra la quota cellulare e il totale [cioè parte cellulare + parte liquida] del sangue), che può raggiungere il 70-80% (valori normali 40-50%).
Come detto, vi può essere un incremento, anche se meno significativo, di globuli bianchi e piastrine.
Per distinguere la PV dalle policitemie secondarie, è utile indagare:
- mutazione di JAK2, presente nel 95% dei casi di PV;
- eritropoietina, che sarà diminuita nella PV e aumentata nelle forme secondarie;
- saturazione arteriosa dell'ossigeno, che sarà normale nella PV e bassa nelle forme secondarie.
La biopsia del midollo osseo non è eseguita di routine nella PV, ma può rendersi necessaria per la diagnosi differenziale con le altre sindromi mieloproliferative o per monitorarne l'evoluzione e eventuali complicanze.
Terapia e prognosi
La PV è una patologia ad evoluzione lenta e, considerando che i pazienti sopravvivono mediamente circa 10-15 anni dalla diagnosi, può essere considerata una delle neoplasie con la prognosi migliore.
Tuttavia, le terapie convenzionali non sono in grado di guarire la malattia e quindi il trattamento deve mirare al controllo della sintomatologia e alla prevenzione delle complicanze trombotiche.
In una minoranza di pazienti, la PV può evolvere in patologie più gravi, come la fibrosi midollare, o trasformarsi in una leucemia acuta.
Nelle fasi iniziali della malattia, per ridurre l'eccesso di globuli rossi, si praticano dei periodici salassi (detti anche "flebotomie"), ovvero si prelevano delle cospicue quantità di sangue, con cadenza variabile a seconda del valore dell'ematocrito.
Al fine di ridurre gli eventi trombotici si utilizza la cardioaspirina o, nei casi più gravi, i farmaci anticoagulanti.
Nelle forme più severe o quando il prurito non passa con gli antiistaminici, si ricorre a dei chemioterapici (idrossiurea) oppure all'interferone α.
Se la splenomegalia comporta sintomi invalidanti, può essere praticata la splenectomia, ovvero la rimozione chirurgica della milza.
L'unico trattamento potenzialmente curativo è rappresentato dal trapianto di midollo osseo; tuttavia esso è gravato da pesanti effetti collaterali e da una mortalità a breve termine molto elevata, quindi non è giustificabile in una patologia tendenzialmente cronica e non fatale come la PV. Nella pratica clinica, viene preso in considerazione solo nei rarissimi casi di PV diagnosticata in pazienti giovani.
Infine, per quanto riguarda le prospettive future, la ricerca scientifica è impegnata nella formulazione e sperimentazione di agenti farmacologici in grado di inibire la chinasi JAK2.
Ultimi articoli sezione: Salute
Molto spesso si sente parlare di farmaci di automedicazione, ma cosa si intende esattamente con questa espressione piuttosto generica?
Come creare un ambiente domestico favorevole al benessere psicofisico
I benefici del tè verde sull’organismo sono noti fin da epoche antiche: possiede proprietà antiossidanti, antinfiammatorie e diuretiche.
In questo articolo abbiamo deciso di raccogliere gli elementi principali da considerare quando ci si trova a prendersi cura di genitori anziani che vivono da soli.
Le intolleranze alimentari sono reazioni avverse dell'organismo provocate dall'ingestione di particolari cibi.
Il vantaggio dell'upscaling: come l'intelligenza artificiale sta migliorando l'imaging medico.
Tra i vari prebiotici disponibili, il lattulosio ha suscitato un crescente interesse per le sue potenziali proprietà benefiche per la salute intestinale.
La tetralogia di Fallot è una malformazione cardiaca congenita complessa, caratterizzata da quattro difetti anatomici.

Accedi ai servizi gratuiti
Se sei già registrato, clicca qui per accedere ai servizi gratuiti:
- Database Alimenti
- Calcola Ricetta
- Slot Machine
Altrimenti, clicca qui per registrarti gratuitamente.
Peste suina: classica, africana, diffusione e prevenzione
Novità da Cibo360 TV

CORSA O PALESTRA PER DIMAGRIRE?

BRUCIA 500 kcal in 30 MINUTI? BALLE!

Qualità delle proteine

Grana Padano o Parmigiano-Reggiano?
